Request More Information
Peptide and Protein Analysis

Every year, the catalog of biomedicines grows broader due to advancements in biomedical research allowing for new drug modalities to be discovered. Fusion proteins, bispecific CAR T-cells, and peptabody therapeutics are all emerging in the biotech market as promising new medicines. In contrast to conventional medicines, referred to as "low molecular drugs," which are produced through chemical synthesis, biomedicines are often produced through a biological synthesis which can add layers of complexity to the process and final product.
Many of them are proteinaceous drugs and antibody drugs developed and manufactured using biotechnologies including genetic recombination, cell fusion, and cell culture. Included in this group is the most widely known biomedicine, the Monoclonal antibody. A mAb is a highly complex biological macromolecule with specific therapeutic effects. It is produced from live cells in extremely complicated culture conditions. Quality control of biopharmaceuticals is a critical step to elucidate any alteration in the primary structure as compared to the reference product. Some of the essential attributes for characterization of mAbs and possible biosimilars include peptide mapping and sequencing analysis of C-terminal and disulfide-bonds linked peptides, glycosylation, and oxidation.
With the high level of complexity in biological synthesis, many analytical techniques are employed to monitor both the progress and final product. Shimadzu offers a wide array of analytical technologies and workflows to enable the bioprocess and bioanalytical scientists to meet their challenges.
Featured Solutions
Glycan Characterization

The majority of proteins synthesized in biological organisms undergo glycosylation. Glycosylation is modification with glycans of high structural heterogeneity composed of multiple monosaccharides, such as glucose and galactose, which are bonded together. These glycans classified into N-linked types and O-linked types. These types of glycan are known to play an important role in regulating protein function, and obtaining information on protein glycosylation is essential to the development of biopharmaceuticals. See how Shimadzu provides a variety of solutions for your glycan characterization needs.
Protein Secondary Structural Analysis by FTIR

X-ray diffraction analysis and NMR are widely used for the structural analysis of proteins. However, infrared spectrophotometry is used for the secondary structural analysis, such as α-helices and β-sheets. As infrared spectrophotometry makes it easy to measure samples in all forms (solid/liquid and crystalline/non-crystalline), it is used to complement the analytical methods mentioned above.
Analysis of S-S Bonds

Introduced here is the analysis using a combination of peptide mapping and mass spectrometry. This method involves digesting the target protein in a proteolytic enzyme such as trypsin under various conditions and comparing the molecular weights of the generated peptide fragments with the theoretical values. The test procedures and results for the analysis of human lysozyme are introduced.
N-Terminal Amino Acid Sequencing of IgG Antibodies

This article introduces an example of amino acid sequencing of mouse antibody IgG using the PPSQ- 51A/53A Protein Sequencer isocratic system as an instance of N-terminal amino acid sequencing of biomedicines.
Featured Applications
LCMS Applications
Characterization of Adeno-Associated Virus (AAV) Capsid Proteins with Liquid Chromatography Mass Spectrometry (LC-MS) Based Peptide Mapping and Post Translational Modification Analysis (PTMs)

In this application, we demonstrate an approach for the characterization of AAV capsid proteins and associated PTMs using a bottom-up method. Digested samples were analyzed using a Shimadzu LC-40 liquid chromatography system coupled with a LCMS-9050 Q-TOF mass spectrometer, providing the necessary sensitivity and mass accuracy to obtain the high sequence coverage. Data acquisition was managed by Shimadzu LabSolutions Software, and data processing was performed on Protein Metrics Software.
Amino Acid Analysis by the Triple Quadrupole LCMS-8060RX

Amino acids are the building blocks of proteins. As such, accurate quantitation of amino acids is essential across many areas in biology, chemistry, and medicine. One of the most powerful tools for amino acid analysis in a variety of matrices is LC-MS/MS; however, quantitation can be challenging as the dynamic range of amino acid concentrations can vary considerably within the same sample. Additionally, reproducibility and robustness for analyses across different matrices can present complications due to variations in ion suppression or enhancement. The new RX source increases reproducibility and robustness while drastically reducing the contributions of unwanted molecules to contamination and matrix effects with the new CoreSpray and improved IonFocus technologies utilizing optimized gas delivery, ESI capillary stabilization, and focusing electrode. This Application News shows accurate quantitation of amino acids across a large calibration range for a mixture of 17 of the common amino acids.
Structural Characterization of Cyclic Peptides using a Quadrupole Time-of-Flight Mass Spectrometer

The aim of this study is structural analysis of a cyclic peptide. We developed an analytical method for cyclosporin A using a LC/Q-TOF mass spectrometer and performed LC-MS/MS measurement. As a result, all of the major MS/MS fragments were assigned to predicted fragments from the chemical structure with a 100% structural coverage. In this presentation, we will discuss the workflow of data analysis using an analytical software and the details of qualitative results. Cyclic peptides are expected to be a new class of therapeutic agents because of their higher stability in vivo and higher cell permeability than linear peptides. MS/MS-based peptide sequencing is the most common effective strategy for structural identification of linear peptides. However, in the case of cyclic peptides, sufficient sequence information cannot be obtained with MS/MS measurement as the fragmentation patterns are quite complicated due to the cyclic nature. In order to overcome this issue, we have attempted to confirm the structural characteristics of cyclic peptides only by spectral assignment.
Direct Quantitation of Intact Proteins By Multiple Ion Chromatogram Method on Q-TOF Mass Spectrometer

With the growth of global protein therapeutic market, it is particularly important to accurately and precisely monitor the protein levels throughout the process of production, characterization, and clinical uses. Over the last 20 years, mass spectrometry has become a powerful analytical technique for protein study. In quantifying intact proteins, selected ion monitoring (SIM) [1] and multiple reaction monitoring (MRM) [2] have been reported by using the triple-quadrupole (QQQ). Quadrupole time-of-flight (Q-TOF) is more sensitive and specific than QQQ, and it is commonly used in profiling analysis. However, due to a lower dynamic range, there is lack of quantitative studies for intact proteins by using Q-TOF. In this study, we aimed to investigate and evaluate the performance of multiple ion chromatogram (MIC) for quantification of intact proteins using Shimadzu LCMS-9030 QTOF mass spectrometer.
A Low Peeling, Fast and Easy Approach for Protein O-linked Glycan Analysis

Protein glycosylation has been shown to be related to protein activity. It is therefore of great interest to study glycan structures of glycoproteins in immunology and cellular biology. On top of that, it is also essential to analyze glycans in recombinant glycoprotein drugs to ensure consistent glycosylation profile. Glycan can be linked to protein either via asparagine (N-glycan) or via serine/threonine (O-glycan). Previously, we have published an application news on N-glycan analysis. 1 Typical analysis of glycan involves cleavage of the glycan from the protein and subsequently derivatization with fluorescent labels (e.g., 2-aminobenzamide, 2-AB) followed by analysis using HPLC. Unlike N-glycans which have well-established glycan releasing strategy, the Oglycans lack practical method for releasing of intact Oglycans. O-glycan releasing method such as hydrazinolysis often results in large amount of side reaction peeling products and requires long preparation time. This study demonstrates a simplified workflow using the S-Bio EZGlycoTM O-Glycan Prep kit, which exhibits low peeling and a reduced sample preparation time for O-glycan characterization. The solution incorporates the Shimadzu Nexera-i MT system and highly sensitive fluorescence detector (RF-20A).
Determination of Molecular Weight of Synthetic Peptides by LC/MS Using Multi-Charged Ion Analysis Software
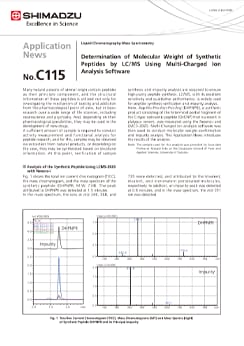
Many natural poisons of animal origin contain peptides as their principle component, and the structural information of these peptides is utilized not only for investigating the mechanism of toxicity and addiction from the pharmacological point of view, but in basic research over a wide range of life sciences, including neuroscience and psychiatry. And, depending on their pharmacological possibilities, they may be used in the development of new drugs. A sufficient amount of sample is required to conduct activity measurement and functional analysis for peptide research, and for this, samples may be obtained via extraction from natural products, or depending on the case, they may be synthesized based on structural information. At this point, verification of sample synthesis and impurity analysis are required to ensure high-purity peptide synthesis. LC/MS, with its excellent sensitivity and qualitative performance, is widely used for peptide synthesis verification and impurity analysis. Here, Asp-His-Pro-Asn-Pro-Arg (DHPNPR), a synthetic product consisting of the N-terminal partial fragment of the C-type natriuretic peptide (OvCNP) that is present in platypus venom, was measured using the Nexera-i and LMCS-2020. Multi-Charged ion analysis software was then used to conduct molecular weight confirmation and impurity analysis. This Application News introduces the results of the analysis.
LC/MS/MS Method Package for D/L Amino Acids

Most important amino acids exist as stereoisomers. D- and L- forms of mirror image isomers, or enantiomers, are named according to their activity on polarized light. By using CROWNPAK CR-I(+) and CR-I(-) columns with chiral stationary phases, the D- and L-forms of amino acids can be analyzed separately. With CR-I(+) elution order is from D- to L-, and with CR-I(-) the elution order is reversed.
High Throughput and High Sequence Coverage Peptide Mapping of a Monoclonal Antibody Using an Integrated Protein Digestion LCMS Platform

This application news demonstrates the advantage of high-throughput and high sequence coverage peptide mapping for the NIST monoclonal antibody (NISTmAb) by coupling an integrated online protein digestion platform, Perfinity Workstation, with a Shimadzu Q-TOF LCMS-9030.
MALDI Applications
Quality Control of Synthetic Peptides using the MALDI-8030 Dual Polarity Benchtop MALDI-TOF Mass Spectrometer

Synthetic peptides are nowadays increasingly used in the fields of biochemistry, immunology and medicine. They fulfil a number of purposes, such as cancer diagnosis and treatment, drug and delivery systems development, epitope mapping, production of antibodies, and vaccine design. The synthesis of peptides is a stepwise process which involves a reaction between the activated carboxylic group of one amino acid and the amino group of another, thus creating the so called peptide bond. The crude peptide product obtained typically contains impurities (e.g., side-products formed during synthesis), hence requiring a purification step. Within the manufacturing process of synthetic peptides, quality control (QC) plays a critical role in providing high purity products. MALDI-TOF mass spectrometry is widely used to confirm the molecular identity of the final peptide as well as its purity. Here, we present the dual polarity MALDI-8030 benchtop linear mass spectrometer for the QC analysis of synthetic peptides in positive and negative ion modes (Fig 1). The benefits of the negative ionisation mode are demonstrated in terms of: i) preserving the integrity of species carrying labile functional groups; ii) simplifying the interpretation of the mass spectra through elimination of salt adduct interferences.
Quality Control Analysis of Synthetic Peptides Using the MALDI-8020 Benchtop Linear MALDI-TOF Mass Spectrometer and QC Reporter Software

Within the manufacturing process of bio-therapeutics, quality control (QC) plays a fundamental role in guaranteeing the supply of a high quality product. Any changes in the product formulation or degradation can affect the therapeutic role, leading to a potential loss of activity or development of toxicity. Amongst the several analytical techniques that can be used to determine the quality of a synthetic bio-product, MALDI-TOF mass spectrometry is widely employed due to its rapid and simple operation, low running costs, sensitivity and ability to provide information on the molecular weight as well as the sequence and structure of a compound, its impurities/adducts and modification products. An example of a modification that can occur as result of degradation is the oxidation of methionine residues of peptides or proteins as this amino acid is highly susceptible to oxidation. Here, we provide a complete workflow solution for the high throughput, automated quality control analysis of synthetic products using the Shimadzu MALDI-8020 benchtop linear MALDI-TOF mass spectrometer and the QC Reporter software.
MALDI-in source decay for the detection of methionine oxidation using the MALDI-8020 benchtop linear MALDI-TOF mass spectrometer

In the pharmaceutical industry, as part of the quality control (QC) process, it is important to track any changes, either due to product formulation or degradation, as they can affect the therapeutic role, leading to a potential loss of activity or development of toxicity. An example of modification than can occur with degradation is the oxidation of methionine residues of a peptide or protein as this amino acid is highly susceptible to oxidation. Here, we demonstrate the capability of the MALDI-8020 benchtop linear TOF mass spectrometer to determine the methionine oxidation (Met-O) of Exendin-4 peptide through accurate intact mass analysis and top-down sequencing. This can be a potentially valuable tool in the QC environments to pinpoint undesired changes during the production process.
Rapid Isolation of Peptides and Proteins from Biological Fluids for Proteomic Analysis by MALDI-TOF Mass Spectrometry

MALDI-TOF mass spectrometry is a versatile analytical technique capable of rapidly characterizing biological samples. The ease of use and high throughput capability make this approach advantageous for analyzing large sample sets. In comparison, the time of data acquisition is significantly less than the initial sample preparation. The isolation, concentration, and buffer exchange necessary for sample preparation can be costly and time consuming. Therefore, new methods need to be developed to isolate and concentrate target molecules quickly and efficiently. In this study, we developed a method to rapidly isolate and concentrate peptides and proteins in biological fluids within a chosen mass range. This method was then applied to mouse plasma, and serum to define a healthy baseline. Proteomic profiling by matrix assisted laser desorption/ionization (MALDI) mass spectrometry (MS) has been shown to be effective at assessing systemic disease1-4, . Other targeted detection technologies such as competitive immunoassays, sandwich-type immunoassays, immune-capture, immunoblots, and activity assays have traditionally been employed to diagnose various disease states. However, MALDI-TOF MS is a sensitive, high throughput, and cost-effective technique that is well suited for analyzing biological fluids. Changes in the relative amounts of peptides and proteins in plasma reflect systemic events; therefore, they can be measured and serve as diagnostic, predictive or prognostic biomarkers of disease. By characterizing these changes in the plasma and serum, insight can be gained into the biochemical events that underpin various disease states.
Protein Identification from Two-dimensional Gel Electrophoresis Based on Peptide Mass Fingerprinting (PMF) Using a Benchtop MALDI-TOF Mass Spectrometer

At present, shotgun proteomics techniques using liquid chromatography mass spectrometry are utilized mainly as high-throughput methods for identifying many different proteins in cellular cytoplasm. However, these techniques are not necessarily effective for identifying all proteins. In particular, when handling proteins separated by means of two-dimensional electrophoresis etc., the protein spots detected on the electrophoresis gel must be linked to the results of protein identification. For such analyses, there may be many cases where using MALDITOF mass spectrometry is more efficient than using liquid chromatography mass spectrometry after enzymetreating the protein spots separated from the gel. This article introduces an example of protein identification using two-dimensional electrophoresis and a benchtop MALDI-TOF mass spectrometer.
Extraction of N-terminally Blocked Proteins from Electrophoresis Gels and Sequence Analysis Using a Benchtop MALDI-TOF Mass Spectrometer

In recent years, it has become possible to analyze the sequence of proteins whose N-terminal is blocked or which are not registered in databases by utilizing protein fragmentation that occurs in the ion source (ISD: In-Source Decay) of MALDI-TOF mass spectrometers. In addition, ISD is theoretically not restricted by the mass of each sample, so high-mass proteins can be sequenced directly without trypsin digestion. Nevertheless, since ISD requires the target protein to be isolated and purified, protein sequence analysis by ISD has yet to gain popularity with non-experts of mass spectrometry. As a solution to such problems, this article introduces an example of combining protein extraction from electrophoresis gels and the MALDI-8020 benchtop MALDI-TOF mass spectrometer (Fig. 1) to measure molecular weights of N-terminally blocked proteins and perform sequence analysis by ISD.
Combining the powerful capabilities of MALDI-TOF MS (MALDI-8020) and Edman Sequencing (PPSQTM-50A Gradient System) for accurate N-terminal sequence of peptides

Mass spectrometry has become an indispensable tool for researchers looking to sequence peptides. Although effective in many cases, sequencing by In Source Decay (ISD) faces a few challenges its ability to provide reliable sequence information including isobaric amino acids, database dependency and low molecular weight interferences. Traditional Edman sequencing avoids mass dependency and the use of databases by analyzing each amino acid from the N-terminus one at a time in sequence. Unfortunately, Edman has its own limitations in providing high sequence coverage. This technical note investigates the benefits of combining the intact mass and sequencing information from the MALDI-8020 (ISD) with the Nterminal sequence obtained from the PPSQ-50A gradient system (Edman). Using the combined information allows investigators the ability to obtain a more complete picture of their proteins and peptides of interest.
Spectroscopy Applications
Protein Quantitation Using BioSpec-nano

Protein solutions contain various coexisting substances, such as surfactants, reducing agents, and denaturants, which affect quantitative analysis results. Therefore, it is necessary to select an appropriate method for each sample. UV absorption (optical density (OD) at 280 nm) is the simplest way to quantitate the total protein concentration using spectrophotometry. This technique, which is based on the 280 nm UV absorption by aromatic amino acids present in proteins, requires no pretreatment and has easy analysis operations. However, absorbance can differ depending on the type of protein, even for the same concentration, and the presence of light-absorbing substances in the same region, such as nucleic acids, prevents quantitation. In contrast, the bicinchoninic acid (BCA) method is a quantitative method that involves small chromogenic differences between different proteins. The method is based on the principle that Cu(I) ions generated in the presence of proteins turn a violet color when they form coordinate bonds with bicinchoninic acid, and is known to offer high sensitivity over a wide concentration range. Thus, although there are various quantitative methods for proteins, it is required to quantify proteins more simply and in smaller amount in the production of biopharmaceuticals. This article describes an example in which immunoglobulin (IgG) was determined by two methods of OD 280 nm method and BCA method using a BioSpec-nano capable of measuring a micro sample of about 1 to 2 µL.
Evaluation of Amyloid-β Aggregation by RF-6000 Spectrofluorophotometer

Amyloid-β is a peptide that consists of approximately 40 amino acid residues. Amyloid fibrils (fibrous aggregates), which occur as a result of the formation of parallel β sheets by intermolecular association of amyloid-β, are the main component of the senile plaques (amyloid plaques) seen in the brains of Alzheimer’s disease patients. Moreover, the formation mechanism and structure of amyloid fibrils have attracted great interest, as amyloid fibrils are also implicated in other neurodegenerative diseases, including Parkinson’s disease (1). Fluorometric analysis using Thioflavin T (ThT), a fluorescent dye used as a marker for amyloid aggregation, is a suitable technique for evaluating amyloid-β aggregation. In this experiment, the formation process of amyloid fibrils was monitored by measuring the fluorescence intensity of ThT and the Rayleigh scattering intensity of the sample solutions, which is proportional to turbidity, at regular time intervals. Although ThT emits almost no fluorescence when it exists in a free state in a solution, it exhibits extremely strong fluorescence (excitation wavelength: 440 nm, fluorescence wavelength: 482 nm) upon binding with amyloid fibrils.
Additional Applications
Evaluation of Protein Aggregation Under Various Stress Conditions Using the Aggregates Sizer

Biopharmaceuticals have recently gained attention for their specificity in attacking pathogens, relative lack of side effects, and potent effect. However, compared with low molecular weight pharmaceuticals, biopharmaceuticals are more susceptible to stress, and are more likely to aggregate. When a biopharmaceutical aggregates due to stress, this results in a decrease or disappearance of its pharmacological effect, along with the potential for causing serious side effects such as shock symptoms from an immunological reaction. Consequently, a framework is being established that evaluates the stability of biopharmaceuticals in terms of their susceptibility to likely stresses (heat and physical stresses during transport, storage, and at use). Protein preparations are a type of biopharmaceutical and aggregate to form sub visible particles (SVP) in the size range of 0.2 to 10 µm. Problems with conventional methods of evaluating protein aggregates have been the inability to analyze the SVP size range in a single measurement, inability to take measurements while stress is applied, inability to recover samples after measurement, and inability to perform quantitative measurements. The Aggregates Sizer biopharmaceutical aggregation analysis system was developed to overcome these problems. This article describes how we applied heat and physical stress to intravenous immunoglobulin (IVIG), then evaluated aggregate formation using the Aggregates Sizer. We show how different aggregate formation processes and speeds occur based on stress type and stirrer bar material by quantifying aggregates in the SVP size range.
Accelerated Testing of Protein Stability Using the Aggregates Sizer TC (With Temperature Control)

Biopharmaceuticals contact a variety of materials during their production, storage, and transport that include metal, plastic and glass. Protein stability differs depending on the materials it comes into contact with. Although investigations must be performed into materials appropriate for contact with biopharmaceuticals, analyzing the many materials a biopharmaceutical contacts during processing increases costs. Furthermore, several months or more are needed to perform a long-term evaluations of storage stability. Accelerated stability testing of contact materials performed in advance would improve the efficiency of investigations into production processes for biopharmaceuticals. We conducted accelerated stability testing of proteins via the monitoring of aggregate formation during application of physical stress at constant temperature. Three agitator plate materials (PEEK, stainless steel, and glass) attached to the “Aggregates Sizer TC (with temperature control)“, Aggregation Analysis System for Biopharmaceuticals was used for testing. The results we obtained indicated the importance of temperature control for stability testing and suggested different materials have different effects on aggregation.
Measurement of total nitrogen (TN) for protein estimation in pharmaceutical vaccines on the Shimadzu TOC-L with TNM-L

This article introduces the application of Total Nitrogen Measurement (TNM) as a powerful analytical technique for protein estimation in biopharmaceutical products such as vaccines. The study was performed at the Serum Institute of India Pvt. Ltd., Pune. Nitrogen is a fundamental component of amino acids, which are the building blocks of proteins. Measurement of nitrogen content is widely being used for the estimation of proteins and its metabolites in pharmaceuticals, foods, beverages, and plant materials. Nitrogen commonly occurs in two forms-organic and inorganic. The inorganic forms include nitrate (NO3 -), nitrite (NO2 -), and ammonium (NH4 +). In pharmaceutical vaccine production, the quantity of the antigen needs to be controlled at the initial-, the intermediate-, and the final stages of the production cycle. Thus, viral vaccines or bacterial vaccines are tested for the quantity of respective attenuated or inactivated viruses or bacteria. Since these antigens typically consist of proteins, analytical quantification of total protein becomes crucial. Diphtheria and tetanus toxoids combined with pertussis antigens are used as a combination DTP vaccine. It confers immunity in children against diphtheria, tetanus, and pertussis. A conventional Kjeldahl method is used for protein nitrogen estimation in vaccines. Its major limitation is the need of different correction factors for different proteins as proteins have variable amino acid sequences. In addition, it is time-consuming and laborious. Further, the use of concentrated sulfuric acid at high temperatures can pose a considerable hazard to safety and health. In this application, high temperature combustion coupled with chemiluminescence detection technique was explored for the estimation of organic nitrogen and inorganic nitrogen. This method provides a fast and an efficient way to monitor nitrogen loading by Total Nitrogen (TN) analysis [1]. The results were compared with both experimental values (Kjeldahl method) and theoretical values.





News / Events
-
AAPS Pharm Sci 360 2025
November 9-12
Henry B. Gonzalez Convention Center
San Antonio, TX -
Pharma Community Network Event
Shimadzu Scientific Instruments and ZefSci invite you and your analytical teams to a pharma community networking event to celebrate 150 years of science and innovation at The Foundry & Lux in South San Francisco. Our event will be built around innovation, collaboration, and connection.
-
ASMS (American Society for Mass Spectrometry) 2025
June 1-5
Baltimore Convention Center
Baltimore, MD -
Shimadzu Scientific Instruments Opens Boston Location of Its R&D Center Focus will be on promoting customer-oriented development to expand business in the pharmaceutical field
Shimadzu Scientific Instruments, Inc. (SSI, Columbia, Maryland, USA), a Shimadzu Group company, has opened a satellite lab in Boston, Massachusetts to be the base of its collaborative research and development activities on the East Coast. Established to conduct research and development more closely linked to customers, SSI's R&D Center consists of three bases, with the main facilities at its Maryland headquarters, a West Coast location, and this new space in Boston near the city center. The Boston lab was set up by partnering with Labshares, a shared laboratory service provider for life science companies.